Nov 1, 2023 - Health
FDA panel weighs CRISPR-based sickle cell therapy

Add Axios as your preferred source to
see more of our stories on Google.
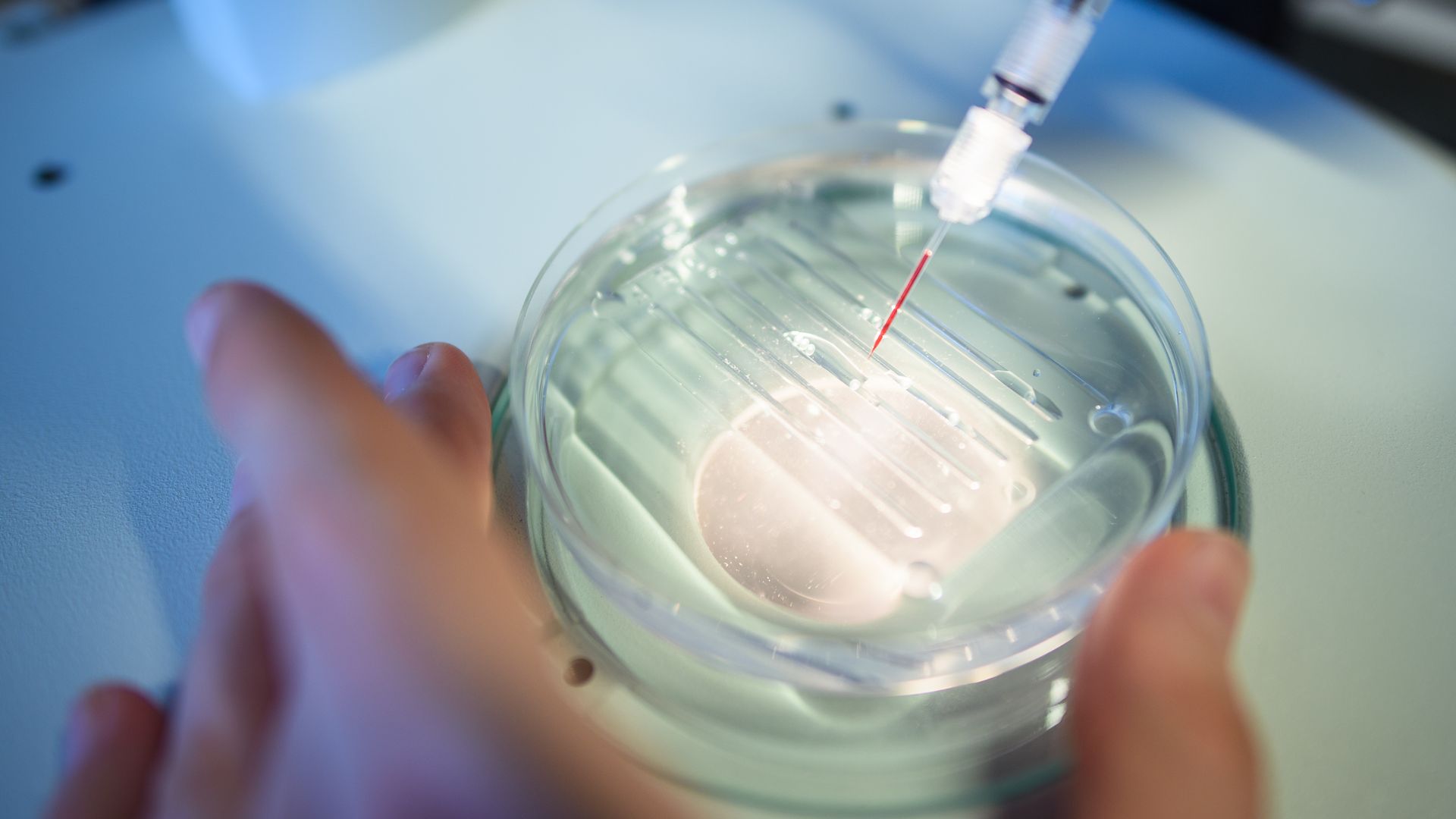
A researcher performs a CRISPR process at the Max Delbrück Center for Molecular Medicine. Photo: Gregor Fischer/dpa
